报告详情
基于深度矩阵分解的空间表观基因组数据去噪
编号:70
访问权限:仅限参会人
更新:2026-03-23 17:20:37
浏览:136次
口头报告
报告开始:2026年03月27日 15:10(Asia/Shanghai)
报告时间:20min
所在会场:[S2] “一作面对面”论坛(信息) [s2] “一作面对面”论坛(信息)
暂无文件
摘要
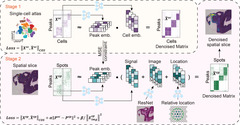
Spatial epigenomics (SE) technologies profile epigenomic landscapes within intact tissues, preserving spatial context and enabling the study of gene regulatory mechanisms in situ. However, current SE datasets typically suffer from low signal detection, substantial noise and extremely sparse peak matrices, which pose considerable challenges for downstream analysis. Here we introduce SPEED (spatial epigenomic data denoising), a deep matrix factorization framework that leverages atlas-level single-cell epigenomic data and spatial context to impute and denoise SE data. In comprehensive benchmarks on both simulated data and real SE tissue datasets, SPEED outperformed five state-of-the-art methods across diverse tissues and technologies. Moreover, SPEED’s denoised outputs facilitated downstream analyses such as differential chromatin accessibility analysis, epigenomic spatial domain identification and gene activity inference. Collectively, our results indicate that SPEED is a generalizable tool for improving data quality and biological insights in SE.
关键词
Spatial epigenomics;Deep matrix factorization;Denoising;Epigenomic spatial domain
报告人

王姝妍
中国科学技术大学稿件作者
全部评论
重要日期
-
会议日期
03月27日
2026
至03月29日
2026
-
03月09日 2026
初稿截稿日期
-
03月29日 2026
注册截止日期
主办单位
中国生物信息学会基因组信息学专业委员会
承办单位
西湖大学
联系方式
- 谭向宇
- 159*********
历届会议
-
2027年05月17日 中国 哈尔滨市(Harbin)
第四届全国基因组信息学大会 -
2025年03月28日 中国
第二届全国基因组信息学大会


发表评论